BIOISIS
It is an open access database dedicated to the study of biological macromolecules by small angle X-ray scattering (SAXS).
The project is supported by the Department of Energy Office of Science Integrated Diffraction Analysis Technologies, the National Cancer Institute Structural Cell Biology of DNA Repair Machines and the National Institute of General Medical Sciences project MINOS (Macromolecular INsights Optimized by Scattering).
BIOISIS aims to become the complete source for the deposition, distribution and maintenance of small angle X-ray scattering data and technologies.
BIOISIS was built with the the Ruby on Rails framework and the MySQL relational database. Its chief designer and architect is Robert P. Rambo, Ph. D., of the Lawrence Berkeley National Lab. The need for a SAXS database for biological sciences was pursued by John Tainer, Ph.D., of the Scripps Research Institute in La Jolla, CA., nd Greg Hura, Ph.D., of the Lawrence Berkeley National Lab in Berkeley, CA.
The database is designed around the concept of an “experiment” and relates a specific experiment to a set of genes, organisms, computational models and experimental data.
Table_organization
If we consider the fact that the structure of a macromolecule in solution is condition specific, then the data and models from a refined SAXS experiment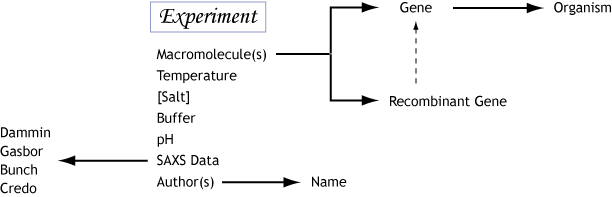
will reflect a condition specific conformational state. Since SAXS experiments at synchrotron sources are highly efficient, a typical data collection at a synchrotron may sample several different experimental conditions, thus yielding measurements of different conformational states. This was an important realization incorporated into the design of BIOISIS.
In contrast, those structures obtained from X-ray crystallography often measure a single conformational state due to both the nature of crystallography and the use of cryogenic temperatures during the experiment.
Latest Journal Articles
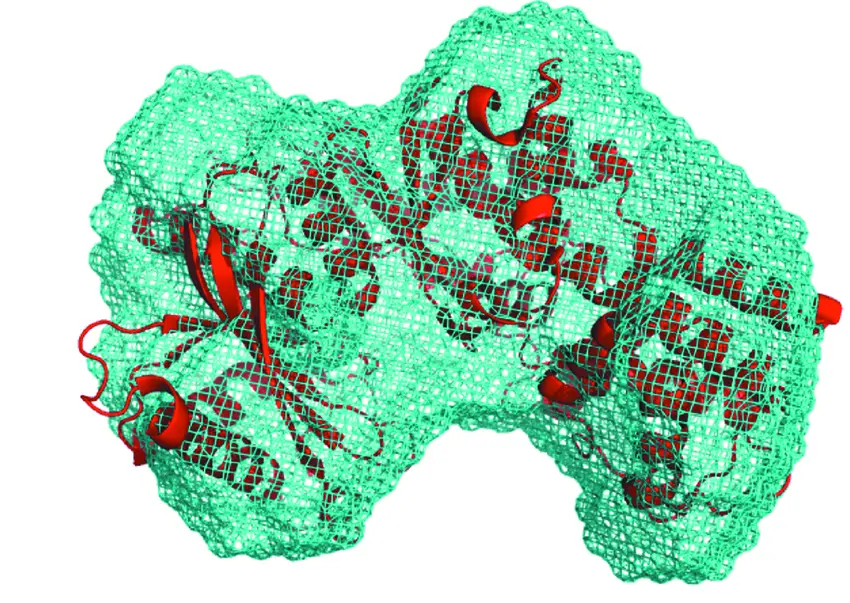
DENFERT: BEAD MODELING WITH HYDRATION
04 September 2014
New dummy atom modeling algorithm that uses the Debye equation instead of spherical harmonic expansions.
The algorithm attempts to model the hydration layer assuming a biphasic composition of the scattering volume.
Incorporation of a hydration layer in the `dummy atom’ ab initio structural modelling of biological macromolecules.
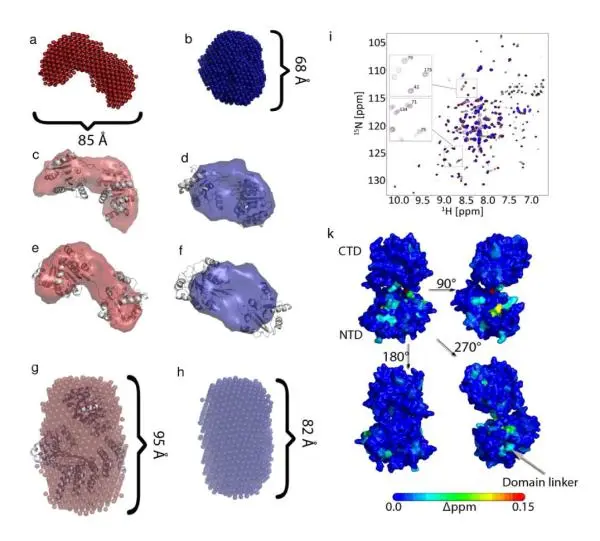
IMPACT OF MACROMOLECULAR CROWDING ON DNA REPLICATION
26 March 2013
Novel application of SAXS using changes in the radius-of-gyration as a function of crowding agent to follow the compaction of an enzyme (in this case the large DNA replication machinery). These types of SAXS studies can be applied to other systems and highlight the effects of modulating water activity on macromolecular structure.
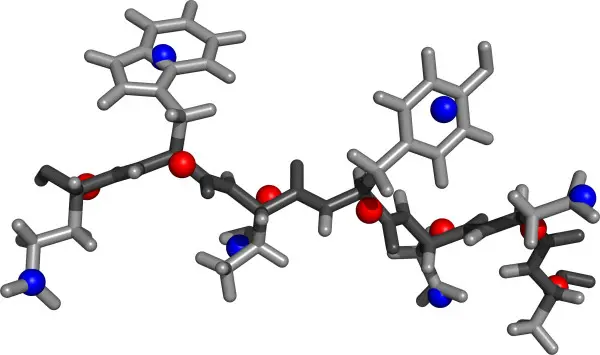
NOVEL 2-BODY COMPUTATIONAL APPROXIMATION FOR SAXS CALCULATIONS
15 January 2011
Excellent computational approach for calculating SAXS profiles from proteins using a 2-body model per amino acid. The authors show, to high scattering angles, the new method is accurate and similar to CRYSOL.
Calculation of Accurate Small Angle X-ray Scattering Curves from Coarse-Grained Protein Models.
BMC Bioinformatics. 2010; 11: 429. Stovgaard K, Andreetta C, Hamelryck T.
News and Updates
09 June 2016 SCATTER 3.0
| ||
|---|---|---|
27 February 2015 RECONSTRUCTION OF SAXS PROFILES FROM PROTEIN STRUCTURES
| ||
04 September 2014 SCATTER 2.2B RELEASED
| ||
24 July 2013 TUESDAY MORNING AT ACA 2013
|