Experimental SAS Curve | |
| ![]()
|
| Experimental Details for BID: TAPSNP |
|---|
| Experiment ID: 114 |
|---|
| Collected at: 18ID/APS (BIO-CAT) |
|---|
| Contributors: Jiang, J , Natarajan, K , Margulies, DH |
|---|
| | Human TAPASIN and TAPBPR (TAP Binding Protein, Related) are homolog proteins. TAPASIN is a part of the classical MHC-I peptide-loading complex (PLC), but TAPBPR is not a component of the PLC. Both interact with MHC-I molecules, but the functionality is different. Results of site-directed mutagenesis of HLA-A2 and TAPBPR are consistent with a view that TAPASIN and TAPBPR share a similar mode of MHC-I binding. The coordinates model of TAPASIN is available from the complex structure of TAPASIN with ERp57 (PDB ID: 3F8U), but the crystallization of TAPBPR and its complexes is difficult. We determined the low resolution structure or “shape” of the TAPASIN and TAPBPR by SAXS studies, and compared the structural similarity and differences. |
|---|
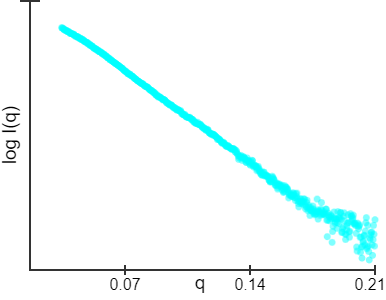
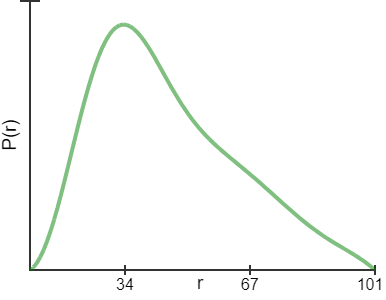
| At BioCAT (18ID/APS) beamline, a size-exclusion chromatography method for data collection was used. The concentration of the loading sample is about 5mg/ml. Purified TAPASIN protein was run in line on a Superdex 200 Increase 10/30 column at a flow rate of 0.75 ml/min directly connected to the sample chamber, and the scattering data were collected with one second exposure per frame during the sample flow through. In this way, the possible aggregation is removed instantly. Two-dimensional diffraction images were reduced to one dimension scattering data and buffer scattering was subtracted. Radius of gyration (Rg), Guinier plot fit and pair-wise distance distribution P(r) calculations were performed using PRIMUS. The dummy residue models were produced using DAMMIN/DAMMIF. The envelopes were reproduced by pdb2vol of SITUS and rendered by VMD. A comparison of Rg and fitness between pdb structures and derived models and the SAXS experimental data were made with CRYSOL. |
|---|
|
|
|---|
| |
|
|
|
|---|
|
|
|
|
The Guinier plot is used to estimate the radius of gyration, Rg, which is taken from the slope of a line observed at low scattering angles (typically in the range where q* Rg < 1.3). This should be in reasonable agreement with the real space Rg. |
|
|---|
| |
| |
The Kratky plot can be used to visually assess the degree of "unfoldedness" of a protein or RNA sample. The plot of a well-behaved folded protein approaches the baseline at high q values creating a parabolic shape. |
|
|---|
| |
| ![]()
|
The red line is the calculated SAXS profile from a PDB model scaled to the experimental SAXS curve (cyan). The two curves agree with a Chi2 of 3.1. |
|
|---|
| |
DAMMIN/F result was determined with the following: | Space Group | P1 | | NSD | RMSD | 0.584 | | variance(NSD | RMSD) | 0.025 | | Number of Models in Average | 10 |
|
|
|
|---|
| Additional Experimental Details |
|---|
| Title TAPASIN (TAP-associated glycoprotein) |
|---|
| Description Human TAPASIN and TAPBPR (TAP Binding Protein, Related) are homolog proteins. TAPASIN is a part of the classical MHC-I peptide-loading complex (PLC), but TAPBPR is not a component of the PLC. Both interact with MHC-I molecules, but the functionality is different. Results of site-directed mutagenesis of HLA-A2 and TAPBPR are consistent with a view that TAPASIN and TAPBPR share a similar mode of MHC-I binding. The coordinates model of TAPASIN is available from the complex structure of TAPASIN with ERp57 (PDB ID: 3F8U), but the crystallization of TAPBPR and its complexes is difficult. We determined the low resolution structure or “shape” of the TAPASIN and TAPBPR by SAXS studies, and compared the structural similarity and differences. |
|---|
| Publication not yet |
|---|
| Contributors Jiang, J , Natarajan, K , Margulies, DH |
|---|
| Genomics and Proteomics The experiment is composed of a single gene/ORF Abbreviated name: hTAPASIN Annotation: Human TAPASIN, Uni-Prot id O15533 TPSN_HUMAN, re-construct with his-tag | GPAVIECWFV EDASGKGLAK RPGALLLRQG PGEPPPRPDL DPELYLSVHD PAGALQAAFR RYPRGAPAPH CEMSRFVPLP ASAKWASGLT PAQNCPRALD GAWLMVSISS PVLSLSSLLR PQPEPQQEPV LITMATVVLT VLTHTPAPRV RLGQDALLDL SFAYMPPTSE AASSLAPGPP PFGLEWRRQH LGKGHLLLAA TPGLNGQMPA AQEGAVAFAA WDDDEPWGPW TGNGTFWLPR VQPFQEGTYL ATIHLPYLQG QVTLELAVYK PPKVSLMPAT LARAAPGEAP PELLCLVSHF YPSGGLEVEW ELRGGPGGRS QKAEGQRWLS ALRHHSDGSV SLSGHLQPPP VTTEQHGARY ACRIHHPSLP ASGRSAEVTL EVAGLSGPSL EDLVPRHHHH HH |
|---|
|
|---|
| category | amino acid composition(%) |
|---|
| Hydrophobic | I(1.2) V(5.7) L(12.9) M(1.5) A(11.2) G(9.2) P(12.7) |
|---|
| Aromatic | F(2.2) W(2.5) Y(2.0) |
|---|
| Hydrophilic | R(5.7) K(1.7) E(5.7) D(3.0) Q(4.5) N(0.7) H(4.7) S(7.2) T(4.2) C(1.2) |
|---|
|
|
|---|
|